Article Text

Abstract
Pulmonary hypertension in type 1 neurofibromatosis is not well known and was previously attributed to diffuse fibrosing alveolitis and parenchymal tumours. More recently, cases of severe pulmonary hypertension due to pulmonary vasculopathy have been described. Involvement of vascular beds, both large and medium calibre vessels, but not pulmonary vasculature, in type 1 neurofibromatosis is well known. The authors describe two such cases and briefly review the literature. Pulmonary arterial hypertension in neurofibromatosis warrants further studies.
- Von Recklinghausen's disease
- pulmonary vasculopathy
- neurofibromin
- pulmonary arterial hypertension
- pulmonary vascular disease
- genetics
- paediatric cardiology
- paediatric interventional cardiology
- echocardiography
Statistics from Altmetric.com
- Von Recklinghausen's disease
- pulmonary vasculopathy
- neurofibromin
- pulmonary arterial hypertension
- pulmonary vascular disease
- genetics
- paediatric cardiology
- paediatric interventional cardiology
- echocardiography
Introduction
Type 1 neurofibromatosis or Von Recklinghausen's disease is an autosomal dominant disorder resulting from mutations in type 1 neurofibromatosis gene which regulates rat sarcoma system of proto-oncogene. The typical clinical features are well characterised and form the basis for the diagnosis.1 Pulmonary arterial hypertension is very rare, and previously thought to result from pulmonary parenchymal involvement.2 3 Severe pulmonary arterial hypertension with advanced pulmonary plexopathy similar to idiopathic pulmonary arterial hypertension is being recognised now, but is very rare.4–11 The relative roles of interstitial lung disease, vascular remodelling and other yet unidentified factors which lead to severe pulmonary arterial hypertension are still elusive. We describe two such cases who had severe pulmonary arterial hypertension with type 1 neurofibromatosis, where no secondary cause including parenchymal involvement could be found and briefly review the existing information about this entity.
Case 1
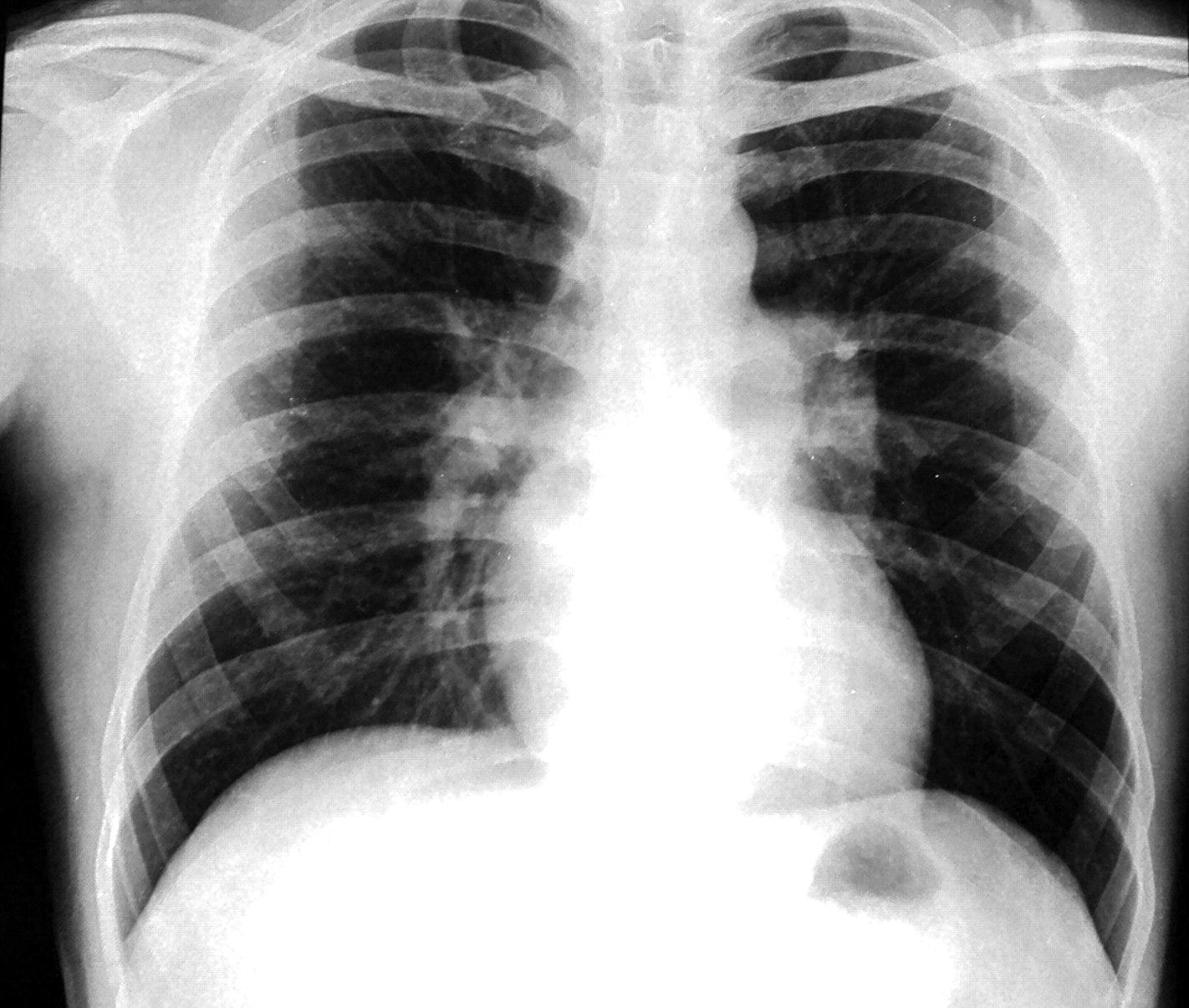
A 34-year-old man presented with 4 months history of progressive exertional dyspnoea. He denied any history of chest pain, orthopnoea and paroxysmal nocturnal dyspnoea. There was no history suggestive of deep vein thrombosis or recurrent thromboembolism, drug abuse or exposure to toxic inhalants or cigarette smoking. Clinical examination revealed tell tale signs of type 1 neurofibromatosis, namely multiple café-au-lait spots, axillary freckling and multiple neurofibromas in upper and lower limbs. His pulse was 110/min regular and blood pressure 110/70 mm Hg. Cardiac examination revealed normal jugular venous pressure and prominent a waves. There was no cardiomegaly. A narrowly split second heart sound with loud pulmonary component was heard on auscultation. He was not in congestive heart failure and other systems including chest were normal. Haemoglobin was 12.3 gm%, and liver and renal function tests were normal. ECG showed normal sinus rhythm with right bundle branch block. Chest x-ray (figure 1) revealed normal cardiac size, enlarged main and right descending pulmonary arteries with pruning and normal lung parenchyma. Echocardiography showed dilated right atrium and pulmonary arteries, mild tricuspid regurgitation with velocity of 4.7 m/s and severe pulmonary arterial hypertension with normal biventricular function. Doppler study of lower limb was normal. High resolution CT was remarkable for bilateral mosaic pattern of ground glass haziness and CT angiography of pulmonary artery showed no thrombi. The patient's haemodynamic data are shown in table 1. He was started on sildenafil 75 mg/day in three doses. Later he underwent a balloon atrial septostomy for two episodes of syncope. He was in New York Heart Association class II status till last follow-up at 11 months.
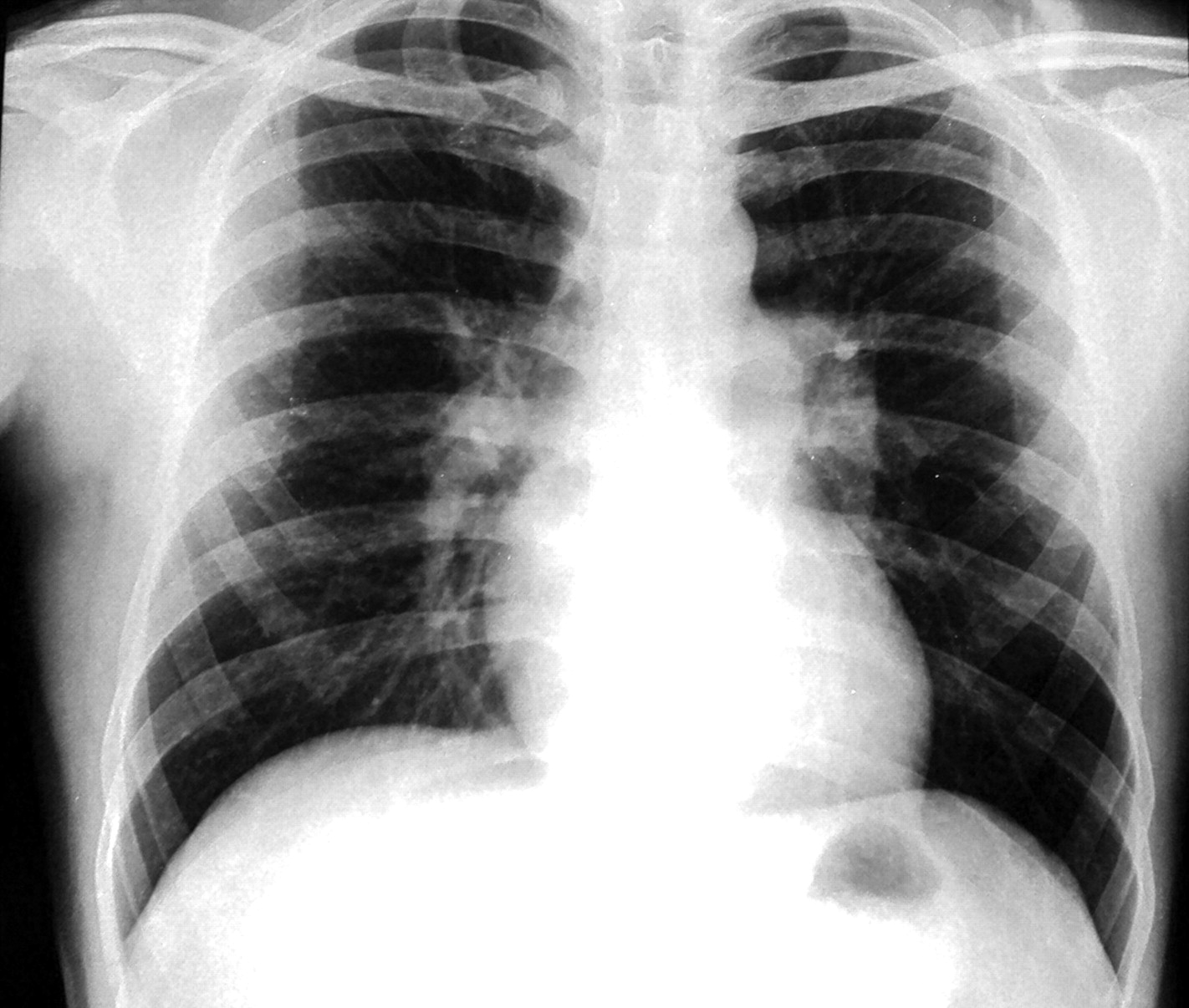
X-ray chest demonstrating prominent central pulmonary artery with peripheral pruning. The parenchyma is normal.
Cardiac catheterisation data
Case 2
A 44-year-old man diagnosed to have type 1 neurofibromatosis since childhood presented with progressive exertional dyspnoea and angina (atypical) on exertion with recent worsening from last 4 months. He was in New York Heart Association class IV status at presentation. There was no evidence to suggest secondary causes of pulmonary arterial hypertension. On examination, heart rate was 125/min and respiratory rate of 23/min with a blood pressure of 90/60 mm Hg. General examination showed café-au-lait spots on trunk, multiple neurofibromas on right lower limb and bilateral pitting pedal oedema. Cardiac examination revealed raised jugular venous pressure with prominent CV waves. There were cardiomegaly, S1 normal, loud P2 with wide fixed splitting and right ventricular S3. Chest and other system examinations were only remarkable for mild hepatomegaly. Laboratory results showed normal blood counts, blood urea 48 mg/dl and creatinine 1.4 mg/dl. Other parameters were normal. ECG showed right ventricular hypertrophy. Chest x-ray revealed cardiothoracic ratio of 65%, and enlarged main and right descending pulmonary arteries with pruning. Echocardiography showed dilated right atrium and enlarged right ventricle pulmonary arteries, moderate tricuspid regurgitation with velocity of 4.3 m/s and severe pulmonary arterial hypertension. Moderate right ventricular dysfunction was present. Doppler study of lower limb was normal. High resolution CT was remarkable for markedly enlarged central pulmonary arteries, irregular opacities with wall thickening, calcification along arterial wall, bilateral mosaic pattern of ground glass haziness and a small healed fibrotic lesion in apical segment of left upper lobe. CT angiography of pulmonary artery showed no thrombi (figure 2). Haemodynamic data are shown in table 1. He was admitted in cardiac intensive care unit and was started on standard heart failure treatment along with bosentan and sildenafil. He improved and was discharged in class II status. Pulmonary function tests revealed forced vital capacity 74% of predicted, forced expiratory volume 66% of predicted, forced expiratory volume to forced vital capacity ratio 92% of predicted and diffusion capacity of carbon monoxide 44%. He was last followed up at 15 months of initial presentation and was in class III status.

High resolution CT of chest depicting bilateral mosaic pattern with variable attenuation and dilated central pulmonary arteries with peripheral pruning.
Discussion
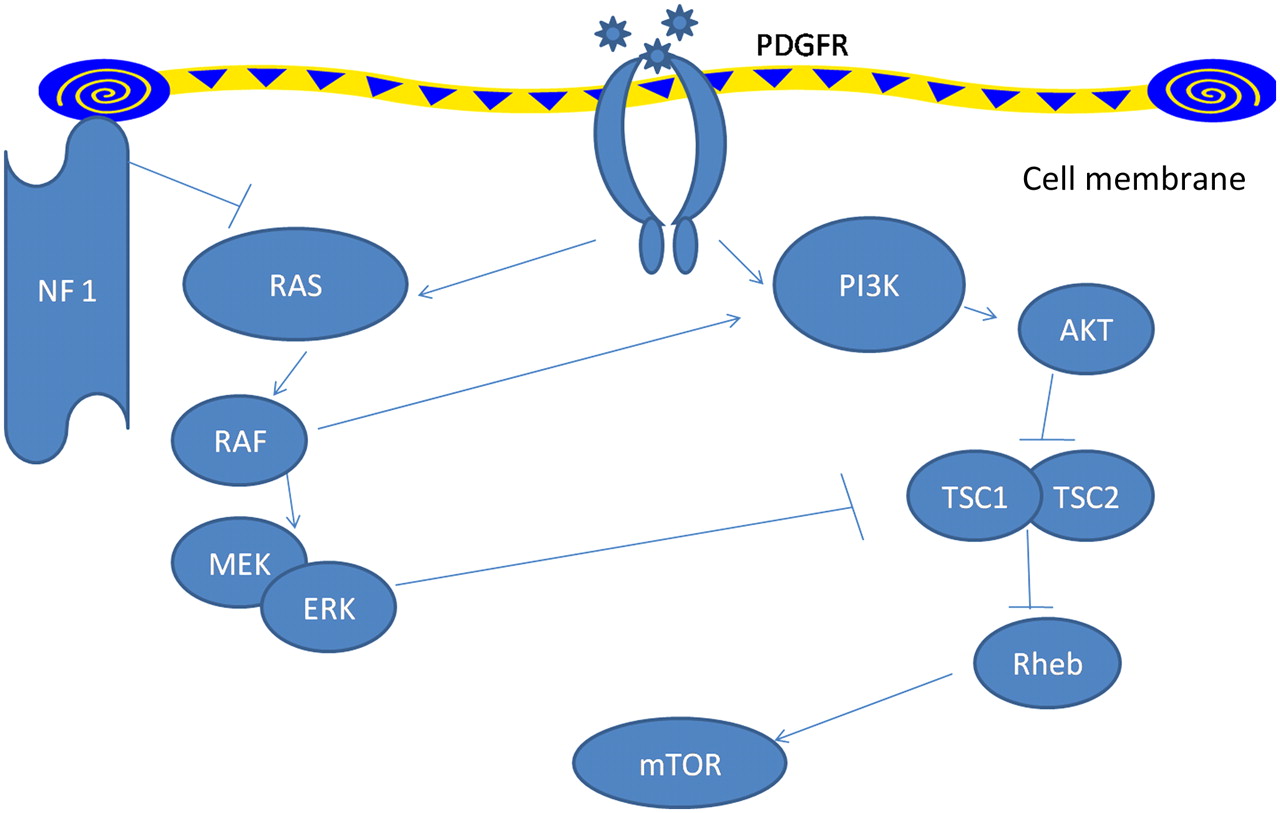
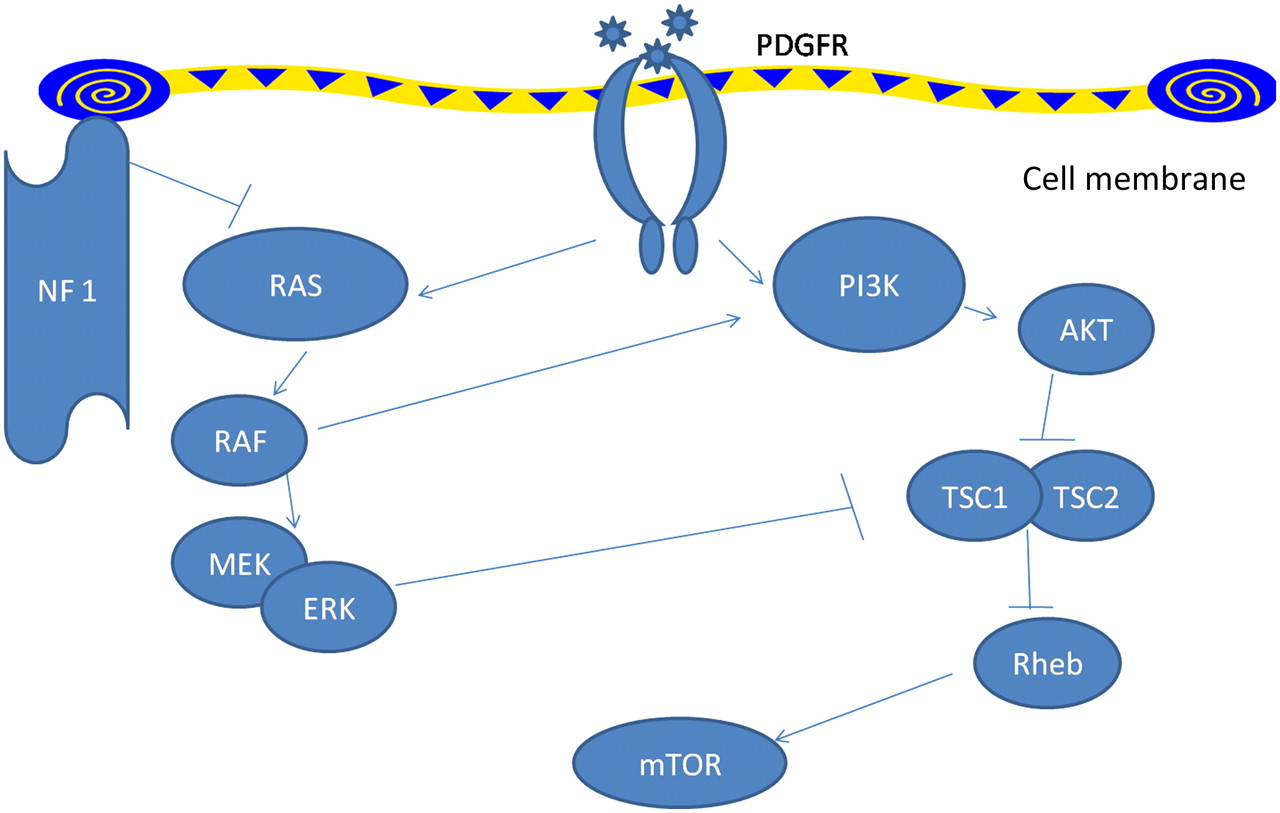
Pulmonary arterial hypertension in type 1 neurofibromatosis has not received enough attention. Recent classification of pulmonary hypertension12 places type 1 neurofibromatosis associated pulmonary hypertension in category 5.2, that is, systemic disorders with unknown or multifactorial aetiology. Initially pulmonary arterial hypertension in type 1 neurofibromatosis was ascribed to severe interstitial fibrosis.2 3 Later, many cases were described where no such associations could be defined and pulmonary arterial hypertension was hypothesised to have resulted from plexiform vasculopathy.4–11 In fact, characteristic mosaic pattern on tomographic scan of chest with histopathological changes of pulmonary vasculopathy with an intimal proliferative plexiform lesions characteristic of type 1 neurofibromatosis arterial involvement4–7 were described and in other cases indirect evidence from imaging and pulmonary function testing was used.8–11 The pathologic picture is remarkably similar to idiopathic and heritable pulmonary arterial hypertension which are also characterised by pathological lesions affecting the pulmonary arteries, distal in particular. They are characterised by medial hypertrophy, intimal proliferative and fibrotic changes (concentric, eccentric), adventitial thickening with moderate perivascular inflammatory infiltrates, complex lesions (plexiform, dilated lesions) and thrombotic lesions. Pulmonary veins are classically unaffected.13 The basic molecular mechanisms by which deficiency of neurofibrin, which has guanosinetriphosphatase activating domain, results in vascular proliferative response is well studied (figure 3). Neurofibrin negatively regulates the Rat sarcoma system of proto-oncogene output; its deficiency causes uninhibited proto-oncogene activity and downstream activation of cell growth. Several pathways, including the mitogen-activated protein kinase pathway and tuberous sclerosis protein 1–2 complex,14 are involved resulting in downstream activation of mammalian target of rapamycin. This in turn increases vascular endothelial growth factor and hypoxia induced factor 1 production.15 16 Neurofibromin also modulates adenylate-cyclase activity through a Rat sarcoma system of proto-oncogene independent mechanism.17 In type II neurofibromatosis manifestations are usually limited to nervous system tissue and no vasculopathy is described. This in turn might be related to the Merlin, the defective gene product which acts through entirely different pathway and target genes might not be expressed in pulmonary vessels. However, the exact mechanism still remains elusive. Table 2 summarises all available case reports and series in type 1 neurofibromatosis patients with pulmonary hypertension.
{kind=link}
{kind=link}
{kind=link}
Signalling pathways modulated by NF1. NF1 leads to decreased activity of Ras thus leading to downstream activation of the vascular growth pathway in NF1-mutated cells. mTOR, mammalian target of rapamycin; NF1, neurofibromin type 1; PDGFR, platelet-derived growth factor receptor; PI3K, phosphatidylinositol 3-kinase; Rheb, Ras homologue enriched in brain; TSC, tuberous sclerosis protein. Pointed arrowheads indicate positive regulation while blunt arrowheads indicate negative regulation.
Summary of type 1 neurofibromatosis cases with pulmonary hypertension
Analysis of all these studies reveals few characteristic features of type 1 neurofibromatosis associated pulmonary hypertension. There is female preponderance unlike type 1 neurofibromatosis itself which affects male and female subjects equally, suggesting a role of oestrogen as reported in heritable pulmonary arterial hypertension.18 In contrast to heritable pulmonary arterial hypertension, the age of onset is very late, with median age being more than 60; also, the median time from diagnosis of type 1 neurofibromatosis to diagnosis of pulmonary arterial hypertension is more than 30 years. This further suggests that arteriopathy of pulmonary vasculature is late phenomenon in natural history of type 1 neurofibromatosis and develops in the long standing disease. Most reported patients have presented with advanced disease before diagnosis and this may be due to lack of awareness of this association and consequent delay in the diagnosis. As previously discussed, the lung abnormalities are variable. Most patients have an impaired diffusion capacity but other parameters are normal. Impaired diffusion capacity in setting of normal lung volumes suggests vascular involvement; it is quite remarkable because type 1 neurofibromatosis patients with lung disease resulting in pulmonary hypertension will invariably show some obstructive, restrictive or mixed patterns and a male preponderance.19 Ventilation perfusion scans and high resolution CT scans of chest might show bilateral filling defects and mosaic pattern (representing irregular perfusion) which again favours vascular involvement as a primary cause of pulmonary arterial hypertension. In all studies where genetic analysis was done, bone morphogenic protein receptor 2 and activin A receptor type II-like kinase-1 mutations were absent, which are known mutations in idiopathic and heritable pulmonary arterial hypertension. In contrast, heterozygous germ line mutations of the type 1 neurofibromatosis gene were present in all cases and were of different types, including short deletions, nonsense and missense mutations, or complete deletion (rare, gene negative cases). No correlation exists between type of mutation and type 1 neurofibromatosis phenotype or development of pulmonary arterial hypertension. The most definitive evidence comes from the cases where biopsy was done and shows the classical plexiform lesions. Undoubtedly, type 1 neurofibromatosis and pulmonary arterial hypertension are rare but it is very important to recognise this association since it implicates mutations in the tumour suppressor gene type 1 neurofibromatosis with the pathogenesis of a pulmonary vasculopathy. Furthermore, early recognition is important because rapid progression and poor prognosis are reported.6 7 11 Both of our patients had long standing neurofibromatosis with advanced disease at presentation and no secondary causes and evidence of primary vascular involvement could be found as described.
Conclusion
In conclusion, pulmonary hypertension in neurofibromatosis type 1 can occur due to parenchymal lung disease or due to disproportionate involvement of pulmonary arteries, or a combination of both. Pulmonary vasculopathy is a rare complication of neurofibromatosis type 1 and has certain distinguishing features. Increased awareness of pulmonary arterial hypertension in neurofibromatosis is warranted. Pulmonary arterial hypertension from pulmonary vasculopathy in type 1 neurofibromatosis may need to be classified differently.
References
Footnotes
Competing interests None.
Provenance and peer review Not commissioned; internally peer reviewed.