Article Text

Abstract
Objective: To measure the inflammatory and autonomic responses of healthy humans and patients with coronary artery disease to controlled concentrations of two specific components of vehicle derived air pollution, carbon particles and sulphur dioxide (SO2).
Methods: Placebo controlled, double blind, random order human challenge study examining the effects of carbon particles (50 μg/m3) and SO2 (200 parts per billion (ppb)) on heart rate variability (HRV) and circulating markers of inflammation and coagulation in healthy volunteers and patients with stable angina.
Results: In healthy volunteers, markers of cardiac vagal control did not fall in response to particle exposure but, compared with the response to air, increased transiently immediately after exposure (root mean square of successive RR interval differences (RMSSD) 15 (5) ms with carbon particles and 4 (3) ms) with air, p < 0.05). SO2 exposure resulted in no immediate change but a significant reduction in HRV markers of cardiac vagal control at four hours (RMSSD −2 (3.6) ms with air, −7 (2.7) ms with SO2, p < 0.05). No such changes were seen in patients with stable angina. Neither pollutant caused any change in markers of inflammation or coagulation at zero, four, or 24 hours.
Conclusion: In healthy volunteers, short term exposure to pure carbon particles does not cause adverse effects on HRV or a systemic inflammatory response. The adverse effects of vehicle derived particulates are likely to be caused by more reactive species found on the particle surface. SO2 exposure does, however, reduce cardiac vagal control, a response that would be expected to increase susceptibility to ventricular arrhythmia.
- APHEA-2, air pollution and health: a European approach
- CRP, C reactive protein
- HF, high frequency
- HRV, heart rate variability
- LF, low frequency
- PNN50, percentage of successive RR interval differences exceeding 50 ms
- RMSSD, root mean square of successive RR interval differences
- SDNN, standard deviation of normal to normal RR intervals
- air pollution
- inflammation
- autonomic nervous system
- arrhythmia
Statistics from Altmetric.com
- APHEA-2, air pollution and health: a European approach
- CRP, C reactive protein
- HF, high frequency
- HRV, heart rate variability
- LF, low frequency
- PNN50, percentage of successive RR interval differences exceeding 50 ms
- RMSSD, root mean square of successive RR interval differences
- SDNN, standard deviation of normal to normal RR intervals
Despite improved urban air quality, exposure to ambient pollution continues to be consistently associated with daily cardiovascular morbidity and mortality. Rates of death and hospital admission for myocardial infarction, congestive cardiac failure, and cardiac arrhythmia are closely correlated with the concentrations of both particulate and gaseous pollutants.1–4 This association is supported by findings from panel studies showing that the risk of myocardial infarction and cardiac arrhythmia increases within hours to days of pollutant exposure.5,6 In addition, interventions resulting in reductions in ambient pollutants have been associated with prompt declines in rates of cardiovascular death.7,8
Whereas it is easy to understand the effects of air pollutants on patients with lung disease, the pathophysiology underlying the relation between air pollution and adverse cardiovascular events is not immediately apparent and, to show causation, plausible biological mechanisms are required. Two hypotheses have been put forward. Firstly, inhalation of fine particles and pollutant gases may provoke an inflammatory response in the lungs with the consequent release into the circulation of prothrombotic and inflammatory cytokines causing endothelial injury and increasing the risk of atherosclerotic plaque rupture and thrombosis.9 Secondly, exposure to pollutants may stimulate airway receptors resulting in an adverse effect on cardiac autonomic control and an increased risk of arrhythmia.10 These hypotheses have been supported by observational community studies, animal work, and a small number of human experimental studies examining responses to concentrated ambient particles and diesel exhaust.11–14 The precise nature of the component pollutants that may be responsible for these effects remains unknown.
In this human challenge study we have measured the inflammatory and autonomic responses of healthy humans and patients with coronary artery disease to controlled concentrations of two specific components of vehicle derived air pollution, carbon particles and sulphur dioxide (SO2).
METHODS
Design
In a random order, double blind, placebo controlled crossover study design, patients with stable angina and healthy volunteers were exposed to controlled concentrations of carbon particles and SO2, alone and in combination, and to medical air as a control.
Exposure system
Pure carbon particles were produced by a spark discharge aerosol generator (GFG-1000; Palas GmbH, Karlsruhe, Germany). The aerosol and air or SO2 were diluted and delivered to a head dome exposure system by a carrier air stream at 120 l/min. Gas flows were controlled by digital mass flow controllers.15 The head dome has been described in detail elsewhere but in brief consists of a transparent acrylic cylinder with a lid, which is lowered over the head of the seated subject.16 The system is sealed by means of a rubber neck seal and gases are vented to the outside.
Pollutant exposures
The doses of pollutants used in this study were selected as being broadly equivalent to the concentrations measured in urban air on days of high pollution. The carbon particle generator has been extensively characterised and was adjusted to produce a mass concentration of 50 μg/m3.17 The particles produced are predominantly between < 10 and 300 nm in diameter, with a mode at 20–30 nm diameter, comparable with the mode in the particle size distribution in roadside air.18 The highest roadside concentration of elemental carbon measured in a study in London and Birmingham in 2000–2002 was 31.4 μg/m3, averaged over 24 hours,19 and hence hourly average concentrations of the order of 50 μg/m3 are likely.
Bottled SO2 was mixed with medical air and regulated at a dose of 200 ppb, a concentration found during air pollution episodes in urban areas. The same doses were used for the combined exposure to SO2 and carbon. The control exposure was bottled medical air at a flow rate of 120 l/min.
Subjects
Twenty patients with stable angina, multivessel coronary artery disease, and good left ventricular function (ejection fraction > 40%) were recruited from the waiting list for coronary artery bypass grafting. Medication was unchanged through the study period. Twenty healthy volunteers of a similar age were recruited from a local family practice. All subjects were screened to exclude diabetes, asthma, and chronic lung disease (table 1).
Baseline characteristics of the two study groups
Protocol
The study was approved by South and East Birmingham regional ethics committees and subjects provided written informed consent. Subjects attended a preliminary visit to the air pollution laboratory to acclimatise themselves to the exposure system and to be trained to breathe in time to an audio signal set close to each person’s resting respiratory frequency.
Studies started between 8–9 am after a light breakfast. Subjects were asked to avoid alcohol and caffeine for 12 hours before each study. Subjects rested semisupine in a temperature controlled laboratory (23 ±1°C) and a venous cannula was inserted into a forearm vein for blood sampling. A standard three lead ECG signal was amplified, processed (high frequency (HF) signal noise filter > 500 Hz), and digitised at 500 Hz with the use of a National Instruments NB/MI0/16XH/18 analogue to digital converter board (National Instruments Corporation). A continuous arterial pressure signal was obtained with the Portapres device (TNO Biomedical Instrumentation) and was similarly digitised. Respiratory excursion was recorded from the output of a strain gauge attached to an elastic strap around the subject’s chest. All signals were displayed on a personal computer running LabView 5.0 software (National Instruments Corporation).
After the patient had rested for 30 minutes, the baseline blood sample was drawn and after a further 15 minutes, two five minute segments of ECG, respiration, and arterial blood pressure data were recorded during controlled respiration. Subjects were then exposed to the pollutant for one hour.
Further blood samples and two further five minute recordings of the heart rate variability (HRV) data were obtained immediately after exposure and again at four hours. A further blood sample was taken at 24 hours. Studies of the remaining pollutants were repeated in random order seven to 14 days apart with identical protocols.
HRV data analysis
All ECG series were edited to exclude ectopic and artefactual signals. The RR intervals before and after any extrasystoles were replaced by interpolation from the preceding and following sinus intervals. No signal containing > 1% of extrasystoles was used for analysis. R waves were detected by an individually adjusted threshold. From each five minute recording, 256 RR intervals were used for HRV analysis. Where both five minute recordings were suitable for analysis the result given is the mean value.
Time domain
The conventional measures of standard deviation of normal to normal RR intervals (SDNN), root mean square of successive RR interval differences (RMSSD), and percentage of successive RR interval differences exceeding 50 ms (PNN50) were calculated. The RMSSD and PNN50 indices are measures of variability of successive differences in RR intervals and assess HF (beat to beat) variation mediated by the vagus nerve.20
Frequency domain
As described in our previous publications, power spectra were analysed by autoregressive modelling, with a model order between 8–20 selected to minimise the Akaike information criterion.21 Powers at low frequency (LF) (centred at 0.1 Hz) and at HF (respiratory frequency, approximately 0.25 Hz) were determined.
Baroreflex sensitivity
Baroreflex sensitivity was determined by measurement of the α index—the transfer function of variability in the systolic pressure signal to the variability in the RR interval. Data segments were accepted if a coherence of > 0.5 was achieved between RR interval and systolic pressure traces. α-HF represents gain in the vagal limb of the baroreflex.20
High resolution C reactive protein
Serum samples were analysed by a latex particle immunoturbidimetric assay on a Roche Modular P800 analyser.
Coagulation markers
Complete blood count, differential white cell count, and platelet aggregation were measured with an automated analyser (Bayer Advia 120). Samples for fibrinogen and d dimers were processed by an MDA coagulation analyser. Fibrinogen was measured by a modified Clauss method (MDA Fibriquik reagent) and d dimers, with a homogeneous latex particle based immunoassay (BioMerieux MDA d dimer kit).
Statistical analysis
Numerical values in the tables for HRV indices are expressed as median and interquartile range. Baseline values, measured on each exposure day for each subject, were compared by analysis of variance. Significance was defined by p < 0.05.
Changes from the baseline values in HRV indices after each exposure were examined by general linear model repeated measures analysis (SPSS version 11; SPSS Inc, Chicago, Illinois, USA). Within subject factors were the time points at baseline, after exposure, and four hours after exposure. Between subjects factors were the exposure and the subject. In this way the effects of pure air and carbon, pure air and carbon plus SO2, and pure air and SO2 were compared.
Values for SDNN, RMSSD, PNN50, HF power, and LF power were not normally distributed and were therefore logged (base 10) before analysis. In all cases the graphs display change from baseline as the mean and 95% confidence interval.
A further general linear model repeated measures analysis was used to investigate the interactions between air, carbon, and SO2. This second analysis used exposure to carbon, exposure to SO2, subject, and group as between subject factors. Testing the interaction terms in this analysis also allowed us to look for differences in the responses between the healthy subjects and the patients.
Statistical power
Based on previous results, the study was designed to give 80% power to detect a difference of one standard deviation in RMSSD at a significance level of 5%.
RESULTS
Laboratory HRV
Baseline HR, blood pressure, and HRV measures within each group were not significantly different on each exposure day (tables 2 and 3). Baseline RR interval and all measures of HRV were higher in the patients with coronary artery disease, probably as a result of β blockade (tables 2 and 3).
Heart rate variability (HRV) and baroreflex sensitivity (BRS) for 20 healthy volunteers before and after exposures to air and pollutants
HRV and BRS for 20 patients with coronary artery disease before and after exposures to air and pollutants
In healthy subjects, exposure to carbon particles resulted in immediate small increases in RR interval, SDNN, and RMSSD that were significantly different from changes observed after exposure to air alone (table 2, fig 1). LF power was increased compared with air and, although HF power rose similarly, this did not reach significance (fig 2). Blood pressure was unchanged. At four hours after exposure to carbon particles, changes in HRV measures were no longer significantly different from those after air exposures (table 2, figs 3 and 4).

Changes from baseline in time domain values of heart rate variability (HRV) for 20 healthy volunteers immediately after exposure to pollutant or air. Bars show the change in mean and error bars show 95% confidence intervals. *p < 0.05 for change from baseline with pollutant compared with change with air (repeated measures analysis). PNN50, percentage of successive RR interval differences exceeding 50 ms; RMSSD, root mean square of successive RR interval differences; SDNN, standard deviation of normal to normal RR intervals.
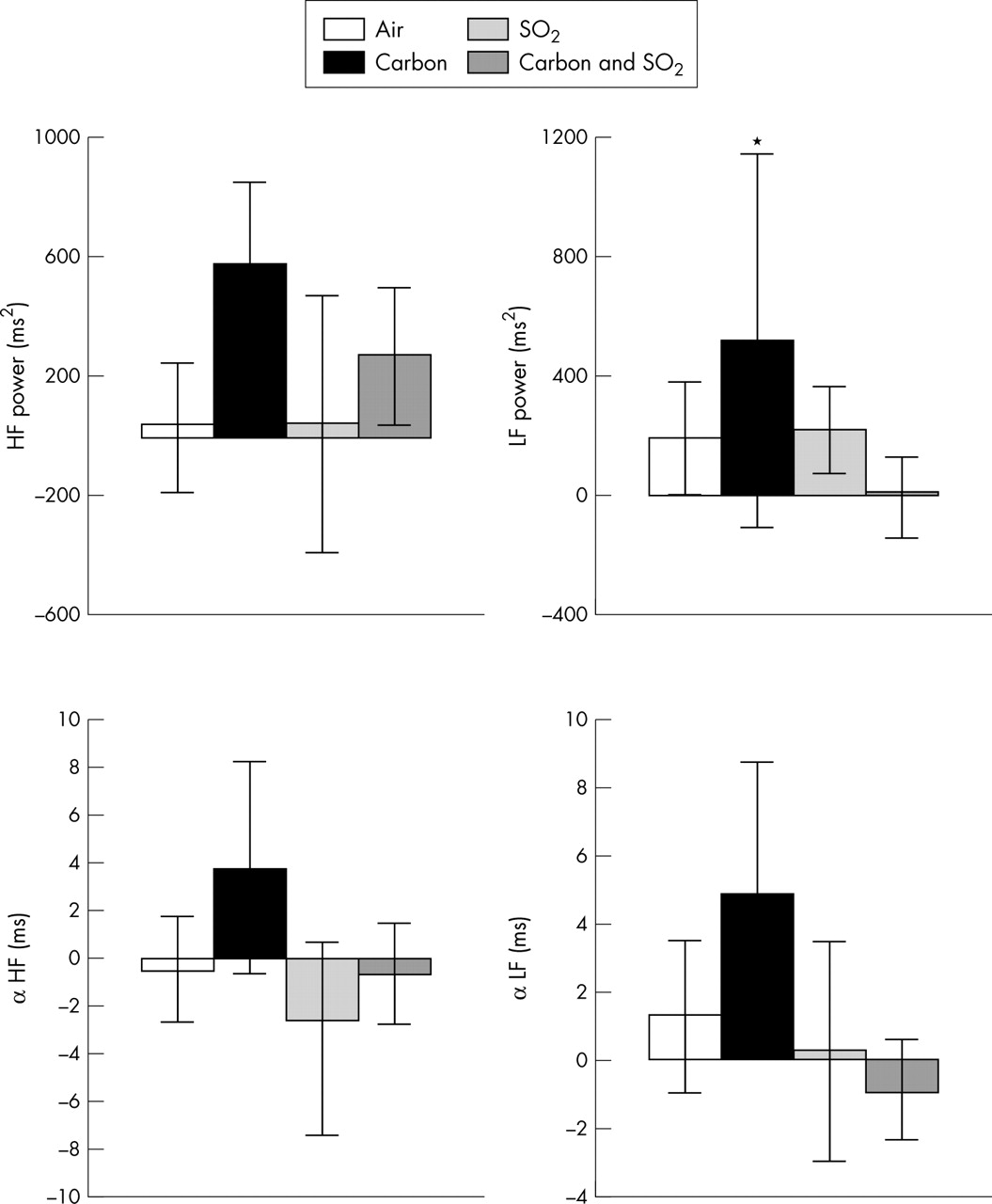
Changes from baseline in frequency domain variables of HRV and baroreflex sensitivity for 20 healthy volunteers immediately after exposure to pollutant or air. Bars show the change in mean and error bars show 95% confidence intervals. *p < 0.05 for change from baseline with pollutant compared with change with air (repeated measures analysis). HF, high frequency; LF, low frequency.

Changes from baseline in time domain values of HRV for 20 healthy volunteers four hours after exposure to pollutant or air. Bars show the change in mean and error bars show 95% confidence intervals. *p < 0.05 for change from baseline with pollutant compared with change with air (repeated measures analysis).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
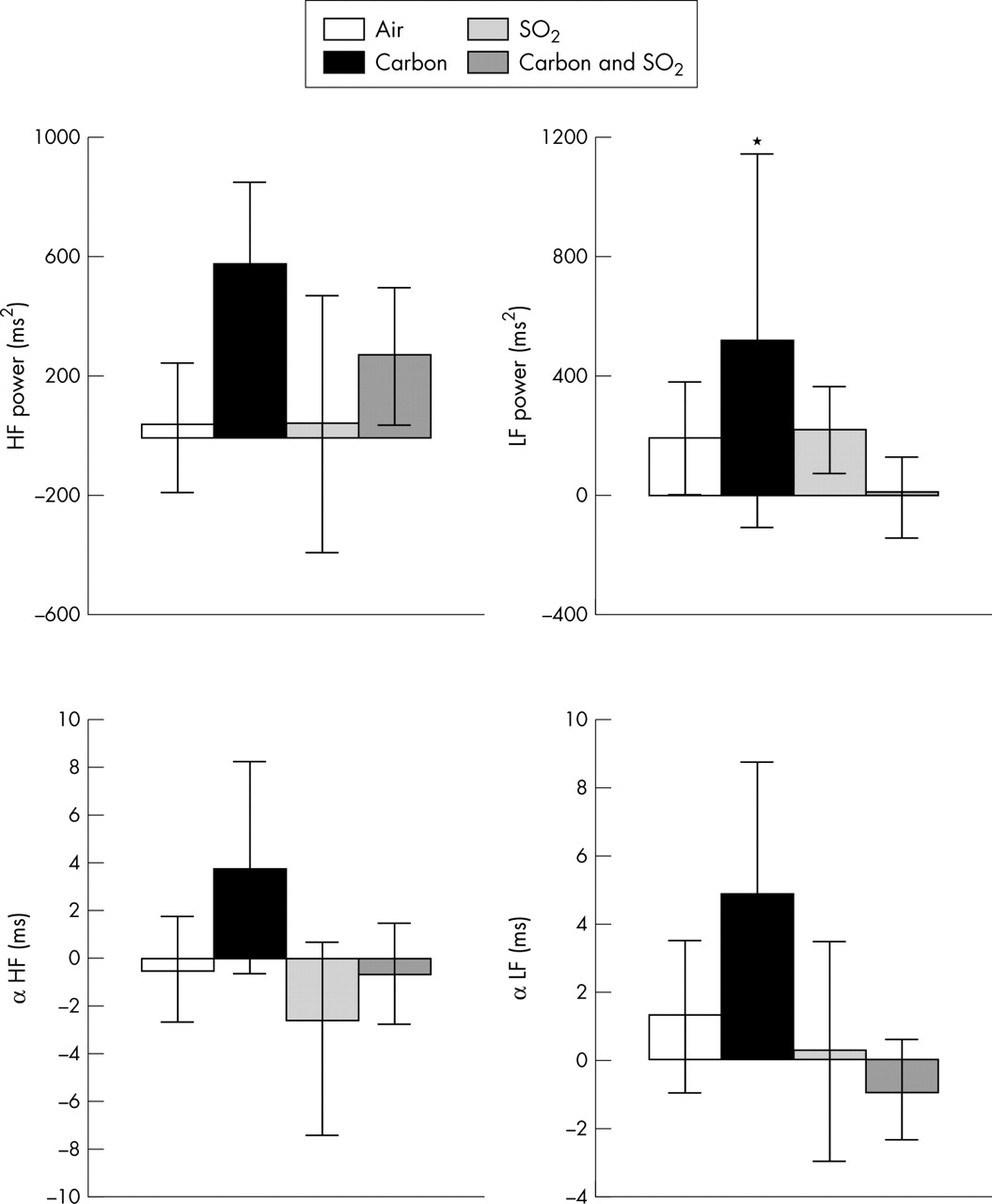
Changes from baseline in frequency domain variables of HRV and baroreflex sensitivity for 20 healthy volunteers four hours after exposure to pollutant or air. Bars show the change in mean and error bars show 95% confidence intervals. *p < 0.05 for change from baseline with pollutant compared with change with air (repeated measures analysis).
Immediately after SO2 exposure HRV did not change significantly. By four hours, however, reductions in RR interval, SDNN, RMSSD, and PNN50 were significantly different from the changes after exposure to air alone (fig 3). HF power changed similarly, although this did not reach significance (fig 4). Changes in α-HF and α-LF after pollutant exposure showed that, compared with air, baroreflex sensitivity was also reduced four hours after exposure to SO2 (table 2, fig 4).
In the initial analysis, the changes in HRV with carbon and SO2 in combination followed a pattern similar to those with SO2 alone but did not reach significance.
The second repeated measures analysis (in which exposure to carbon and exposure to SO2 were entered as between subjects factors) showed a significant interaction between carbon and SO2, such that the effect of SO2 on HRV appeared to be modulated by the presence of carbon (p < 0.001 for RMSSD and p < 0.005 for SDNN).
In patients with coronary artery disease, the changes in HR, blood pressure, and all HRV measures after pollutant exposure did not differ significantly from those after exposure to air (table 3). When the two patient populations were compared by multiple comparison analysis, the group responses to exposure were significantly different with little or no response in the patients with coronary disease (p < 0.001 for RMSSD and p < 0.001 for SDNN).
Complete blood count
Haemoglobin, platelet count or aggregation, or differential white cell count did not change significantly at four or 24 hours after pollutant exposure.
High resolution C reactive protein and coagulation markers
Baseline concentrations of C reactive protein (CRP) were higher in patients with coronary artery disease than in the healthy subjects (3.95 (0.22) v 2.21 (0.35) mg/l, p < 0.01). Baseline high resolution CRP concentrations did not differ significantly between study days. High resolution CRP did not change significantly in response to any pollutant exposure in either the healthy volunteer group or the patient group. Similarly, there was no evidence of a significant effect of any pollutant on concentrations of fibrinogen or d dimer.
DISCUSSION
Heart rate variability
These results suggest that short term exposure to SO2 causes a decrease in measures of cardiac vagal control and baroreflex sensitivity in healthy humans. The effect was observed four hours after exposure to SO2 at 200 parts per billion (ppb), a concentration often encountered during pollution episodes. This result is consistent with the findings of several observational studies such as APHEA-2 (air pollution and health: a European approach), in which daily hospital admissions for ischaemic heart disease were independently associated with changes in daily concentrations of SO2.4 A fall in ambient SO2 in Hong Kong after changes in fuel composition was also associated with a substantial (2%) decrease in annual cardiovascular mortality.8 Indirect support that these effects are mediated by changes in cardiac autonomic control comes from a German panel study, which showed an independent association of heart rate with ambient SO2 concentration.12 The results of our challenge study now provide direct evidence that SO2 exerts autonomic effects that are well known to increase susceptibility to ventricular arrhythmias.10,22
The observed changes in cardiac vagal activity are likely to have resulted from upper airway receptor stimulation. SO2 is a highly soluble gas and at the concentrations used in this study would not be expected to penetrate the respiratory system beyond the trachea.23 SO2 is, however, known to be able to activate rapidly adapting receptors and C fibres in the upper airway and to induce local airway narrowing by stimulating sensory mucosal nerve endings.24 The lack of change in CRP and procoagulant activity supports a direct neural effect rather than effects secondary to a systemic inflammatory response. Further research is required to elucidate the site and time course of SO2’s influence on autonomic control.
We have also shown that exposure to pure carbon particles results in immediate but transient alterations in cardiac autonomic control. The nature of this change in cardiac autonomic control, bradycardia, and increases in HRV measures of cardiac vagal activity is not in accord with the results of observational studies. Most of these suggest that high concentrations of particulate matter are associated with an increase in heart rate and a reduction in HRV.25–27 Furthermore, a recent human challenge study showed that concentrated ambient particle exposure results in an immediate reduction in HRV.13 The apparent discrepancy between these data and our results must lie in the nature of the particles. Whether it is particle size or composition that determines toxicity remains contentious.28 Ambient particles are complex mixtures of elemental carbon, organic molecules, and highly reactive species such as transition metals, which have been implicated as a cause of adverse health effects. This issue was addressed in our study by the use of particles of a size similar to that of the products of vehicle combustion but composed of elemental carbon alone. Our findings would suggest that in humans, pure elemental carbon particles of equivalent size to those derived from combustion exert a small vagal stimulatory rather than inhibitory action. Such an effect is unlikely to predispose to ventricular tachyarrhythmia and sudden death. These results provide indirect evidence that it is the non-carbon content of particles that exerts adverse cardiovascular effects.
The lack of any change in HRV in response to pollutant exposure in patients with coronary artery disease requires explanation. This finding contrasts with the effects observed in normal volunteers and with the widely held view that air pollution predominantly affects high risk people such as those with existing coronary artery disease. We were surprised by this finding but we believe that the different HRV response to exposure between the two groups was a result of drug treatment rather than an inherent difference in susceptibility to the effects of air pollutants. A high proportion of our patients were taking β blockers, which are known to exert powerful effects on HRV, increasing indices of cardiac vagal control.29 In addition we cannot exclude possible effects of statins and aspirin on both HRV indices and inflammatory responses. The potential masking of responses to pollutants by drugs illustrates the difficulty of human research in this area, but drug withdrawal poses ethical dilemmas and arguably makes the experimental protocol less relevant to real life exposure.
Inflammation and coagulation
The adverse influence of SO2 exposure on cardiac autonomic control cannot be attributed to a systemic inflammatory response. None of our pollutant exposures was accompanied by a systemic acute phase or coagulant response. Epidemiological studies have shown a significant association between both SO2 and ambient particle concentrations and values of plasma CRP and viscosity.11,30 As concentrations of these pollutants often rise and fall in parallel, it is difficult to determine the culprit pollutants from such studies. This requires controlled human exposure studies. Exposure of healthy volunteers to diesel exhaust results in pulmonary inflammation and an increase in circulating platelets, neutrophils, and adhesion molecules.14 Similar changes, including an increase in fibrinogen, occur after experimental exposure to concentrated ambient particles.31 The results we present therefore constitute an important negative finding and suggest that the inflammatory response to fine particles is likely to depend on their composition. The presence of reactive species such as transition metals may be necessary to cause adverse cardiovascular effects. Consistent with our results, exposure to pure carbon particles in animal studies does not evoke an inflammatory response.32 Such a response depended on the bioavailability of transition metals.33
Limitations
The lack of significant reductions in HRV in response to carbon particles is an important negative finding. The size of an effect on HRV that could be excluded (with > 80% certainty) according to the statistical power of this study was one of 1.9 times and 1.3 times the change in response to air for HF power and RMSSD, respectively. Clearly smaller changes than this in cardiac autonomic tone cannot be excluded. As there is no definable threshold effect for a change in cardiac autonomic control, even very small reductions in HRV measures of vagal activity may reflect changes sufficient to increase the chances of a susceptible person having an arrhythmic event.
We exposed subjects to pollutants at concentrations similar to those experienced in urban areas and to those that have been associated with mortality and admissions for cardiovascular disease. It was not our intention to investigate the effects of pollutants at high concentrations experienced infrequently during severe pollution episodes but rather to explain the influence that low dose pollutants have on cardiovascular mortality in urban areas from day to day.34 Higher concentrations and more prolonged exposures possibly exert different autonomic and inflammatory effects.13 Our exposure duration was brief and, as our subjects were resting during exposure, the inhaled dose may have been lower than that in an ambulatory setting. Previous exposure protocols have overcome this limitation by incorporating exercise to increase respiratory rate and depth.31 This would, however, have made the recordings of stationary ECG series difficult. Data were collected at isolated time points after exposure and the possibility that the autonomic or inflammatory effect of either pollutant peaked between or after these time points cannot be excluded.
Although this study raises the possibility of an adverse effect of SO2 on cardiac autonomic control we cannot claim to have found strong evidence for an inflammatory or autonomic mechanism by which particulate air pollution increases adverse cardiovascular events.
Alternative hypotheses, such as a direct effect on coronary arteries mediated by particles that may enter the circulation, cannot be excluded, although as yet there is little supportive evidence.35 In contrast there is a growing body of work in support of both autonomic and inflammatory influences of fine particles. Before these hypotheses are rejected, further research is required. Our results suggest that future efforts should be concentrated on possible autonomic effects of SO2 and on the responses to non-carbon components of ambient particulate matter such as transition metals.
Acknowledgments
Statistical advice was supplied Dr PG Nightingale, Wellcome Trust Clinical Research Facility.
REFERENCES
Footnotes
-
Published Online First 27 May 2005
-
This work was supported by a project grant from the British Heart Foundation.