Article Text

Abstract
Objective: To determine the range of survival rates of patients with hypertrophic cardiomyopathy (HCM) by comparing and contrasting the natural history of a cohort of patients seen between 1988 and 2002 with that of other published series.
Methods: 956 adult (⩾ 16 years old) patients with HCM (572 men, mean (SD) age 42 (15) years, range 16–88) were evaluated by ECG, Holter, exercise testing, and echocardiography. Patient characteristics and survival data were compared with those in natural history studies from referral and non-referral centres published between 1960 and January 2003.
Results: The duration of follow up was 69 (45) months. 120 (12.6%) patients died or underwent cardiac transplantation. Sudden cardiac death (n = 48) was the most common mode of death. The annual rate of sudden death or implantable cardioverter-defibrillator discharge was 1.02 (95% confidence interval (CI) 0.76 to 1.26). Annual rates for heart failure death or transplantation and stroke related death were 0.55% (95% CI 0.37% to 0.78%) and 0.07% (95% CI 0.02% to 0.19%), respectively. When studies published within the last 10 years of the study period were compared with earlier reports, the size of individual study cohorts was larger (309 (240.6) v 136.5 (98.8), p = 0.058) and the proportion with severe functional limitation NYHA class III/IV lower (12.4% v 24.8%, p < 0.0001), and fewer patients underwent septal myotomy-myectomy (5.2% v 18.7%, p < 0.0001). Published sudden death rates over the last 10 years were lower than previously published figures (median 1.0% (range 0.1–1.7) v 2.0% (0–3.5)).
Conclusion: Published survival rates in HCM cohorts have improved progressively over the past 40 years. In the modern era the prevalence of disease related complications is similar in all reporting centres.
- CI, confidence interval
- HCM, hypertrophic cardiomyopathy
- ICD, implantable cardioverter-defibrillator
- prognosis
- hypertrophic cardiomyopathy
- referral bias
Statistics from Altmetric.com
Hypertrophic cardiomyopathy (HCM) is a genetically transmitted disease with a variable clinical course and outcome. In many series, annual death rates are as high as 3–6%,1–4 but recent studies from centres in Europe and the USA report a substantially better prognosis with overall mortality of 1% or less.5–9 It has been suggested that these differences in survival are explained by tertiary centre bias, with the higher rates representing the effect of selective referral of high risk patients to specialist units.1,6,10 An alternative explanation is that differences in published survival rates reflect historical trends in treatment and diagnostic algorithms.
Over the past decade, about 1000 patients with HCM have been evaluated at our centre by a standardised approach to diagnosis and treatment. The objective of this study was to compare and contrast the natural history of this cohort with other published series to determine the range of published survival rates in the modern era.
METHODS
Patients
The study cohort comprised 956 consecutively evaluated adults aged 16 years or more (572 men, mean (SD) age 42 (15) years, range 16–88) with HCM assessed in a dedicated HCM clinic at St George’s Hospital, London, UK, between February 1988 and June 2002. Only patients with follow up of six months or more were included in this study. HCM was diagnosed after successful resuscitation from an out of hospital cardiac arrest in 18 (1.9%) patients, after presentation with symptoms in 457 (47.8%), and during familial assessment in 237 (24.8%). The diagnosis was an incidental finding in 244 (25.5%) patients. The reasons for referral to the clinic were clinical management (n = 344 (36.0%)), risk stratification (n = 173 (18.1%)), family screening (n = 158 (16.5%)), diagnostic clarification (n = 105 (11.0%)), and genetic counselling or referral for second opinion (n = 176 (18.4%)).
The diagnosis of HCM was based on the echocardiographic evidence of left ventricular hypertrophy greater than two standard deviations for age or in accordance with criteria for the diagnosis of familial disease in patients with at least one first degree relative with an unequivocal diagnosis.11,12 Patients with other cardiac or systemic diseases that can produce hypertrophy were excluded.
Clinical evaluation
The initial clinical evaluation comprised full history and physical examination, 12 lead ECG, two dimensional/Doppler echocardiography, 48 hour ambulatory ECG monitoring, and symptom limited cardiopulmonary exercise testing with simultaneous measurement of blood pressure in accordance with previously published methods.13–17
Patients with moderate to severe resting left ventricular outflow tract obstruction were given β blockers, disopyramide, or both for symptom control. Patients who remained symptomatic or were unable to tolerate medication were offered septal myotomy-myectomy, pacemaker (DDDR) implantation, or septal alcohol ablation.
All patients underwent risk stratification in accordance with published protocols.18–25
Patients considered to be at risk of sudden cardiac death were offered treatment with amiodarone or an implantable cardioverter-defibrillator (ICD). Amiodarone was used to prevent atrial fibrillation and supraventricular arrhythmia. After loading, amiodarone dosage was adjusted to maintain a plasma concentration between 0.5–1.5 mg/l. Anticoagulation was started for all patients who had had an embolic event and for those with evidence of sustained or paroxysmal supraventricular tachycardia or frequent palpitations in the presence of left atrial enlargement.
Survival analysis
Data on survival and clinical status were collected at clinic visits from patients followed up at this institution and by direct communication with patients and their general practitioners for those followed up elsewhere. The following end points were used in the survival analysis: (1) sudden cardiac death—witnessed sudden death with or without documented ventricular fibrillation, death within one hour of new symptoms, nocturnal death with no antecedent history of worsening symptoms, and successfully resuscitated cardiac arrest; (2) progressive heart failure death—death preceded by signs and symptoms of heart failure or cardiogenic shock; (3) other cardiovascular death—death precipitated by stroke, pulmonary embolism, and myocardial infarction; (4) procedure related death—death caused by or related to surgical or interventional procedures (for example, myotomy-myectomy, alcohol ablation, or device implantation); (5) non-cardiovascular death—death caused by known non-cardiovascular or unknown events; and (6) orthotopic heart transplantation.
For patients with ICDs, the first appropriate shock was coded as an outcome in a separate survival analysis.
Literature search
Published natural history studies to January 2003 were identified by searching the Cochrane and Medline databases. The initial search terms were “hypertrophic cardiomyopathy”, “sudden death”, and “survival”. References from these reports and earlier reviews were also searched manually. Annual sudden cardiac death and all cause death rates were calculated for each study; 95% confidence intervals (CIs) were calculated by using the Poisson distribution.
Data analysis
Data were statistically analysed with SPSS (version 10.0) statistical software (SPSS Inc, Chicago, Illinois, USA). All data are expressed as mean (SD) (range) or frequency (percentage). Student’s t test was used for comparisons between continuous variables and Pearson’s χ2 or Fisher’s exact test was used for dichotomous variables. Survival estimates were calculated by the Kaplan-Meier method. Five year survival values are expressed together with their 95% CI, defined as survival (1.96) × SE. Cox regression analysis was used to investigate the relation between significant variables and survival. Values of p < 0.05 were considered significant.
RESULTS
Clinical course
Table 1 shows baseline clinical and echocardiographic data in the study cohort.
Baseline clinical characteristics of the study population
Follow up duration was 69 (45) (6–172) months. During follow up, 414 (43.3%) patients received β blockers, 267 (27.9%) calcium antagonists, 112 (11.7%) disopyramide, 322 (33.7%) amiodarone, and 209 (21.9%) warfarin. Indications for amiodarone were supraventricular arrhythmia (n = 152 (47.2%)), sudden death prophylaxis (n = 87 (27.0%)), or both (n = 83 (25.8%)). Fifty two (5.4%) patients received an ICD (41 (78.8%) for primary and 11 (21.1%) for secondary prevention) and 110 (11.5%) patients required a pacemaker: 49 (44.5%) for refractory symptoms; 44 (40.0%) for conduction disease; 8 (7.3%) for chronotropic incompetence; 6 (5.5%) for treatment optimisation; and 3 (2.7%) for other reasons. Thirteen (1.4%) patients had undergone septal myotomy-myectomy before their first visit. Twenty seven (2.8%) patients underwent septal myotomy-myectomy and 21 (2.2%) patients underwent septal alcohol ablation during follow up.
During follow up, the majority (n = 808 (84.5%)) of patients remained in NYHA class I or II. One hundred and thirty one (13.7%) patients developed severe dyspnoea (class III to IV) despite medical treatment or intervention. Eighty six (9.0%) patients reported new onset syncope and 195 (20.4%) reported exertional chest pain. Eighty two (8.6%) patients developed persistent or paroxysmal atrial fibrillation. Three (0.3%) patients developed infective endocarditis; all had left ventricular outflow tract obstruction and one required mitral valve replacement.
Survival
During follow up, 120 (12.6%) patients died or underwent heart transplantation (48 sudden cardiac death, 21 heart failure related death, nine heart transplantation, four stroke related death, five procedure related deaths, 12 other cardiovascular death, and 21 non-cardiovascular death). The five year and 10 year cumulative survival rates from all cause mortality or cardiac transplantation were 91.2% (95% CI 89.1 to 93.3) and 80.3% (95% CI 76.2 to 84.4), respectively (fig 1). The five year and 10 year cumulative survival rates from sudden cardiac death or cardiac arrest were 95.6% (95% CI 94.1 to 97.1) and 91.8% (95% CI 89.1 to 94.5), respectively. Eight of 52 (15.4%) patients with an ICD experienced one or more appropriate discharges. When sudden death, successfully resuscitated ventricular fibrillation, and ICD discharges were considered together, the combined annual sudden cardiac death rate was 1.02% (95% CI 0.76 to 1.26).

Cumulative survival rates for all cause mortality or cardiac transplantation, sudden cardiac death, and heart failure death or transplantation.
During follow up, 31 (3.2%) patients (age 47 (16), 20 men) had an embolic stroke or a transient ischaemic event (lasting < 24 hours). The annual rate of stroke was 0.56% (95% CI 0.38 to 0.80). Atrial fibrillation (20 (64.5%) v 137 (14.8%), p < 0.00001) and left atrial enlargement (25 (80.6%) v 316 (35.1%), p < 0.00001) were more common in patients who had a stroke or transient ischaemic event. Stroke was a terminal event in four patients (age 63 (11) (49–71) years).
Comparisons with published survival data
Table 2 shows clinical and survival data from previously published natural history studies.2–9,18–20,24–32 Characteristics of patient cohorts in studies published more than 10 years ago differed substantially from those of recent reports (⩽ 10 years). More recent individual study cohorts were larger (309 (240.6) v 136.5 (98.8), p = 0.058), the proportion with severe functional limitation NYHA class III/IV was lower (12.4% v 24.8%, p < 0.0001), and fewer patients underwent septal myotomy-myectomy (5.2% v 18.7%, p < 0.0001).
Comparisons of clinical features and outcomes in selected natural history studies
Figures 2 and 3 show annual sudden death and all cause death rates, respectively, in each of these series together with their 95% CIs. Annual sudden death rates over the last 10 years of the study were lower than previously published figures (median 1.0% (range 0.1–1.7) v median 2.0% (range 0–3.5)).
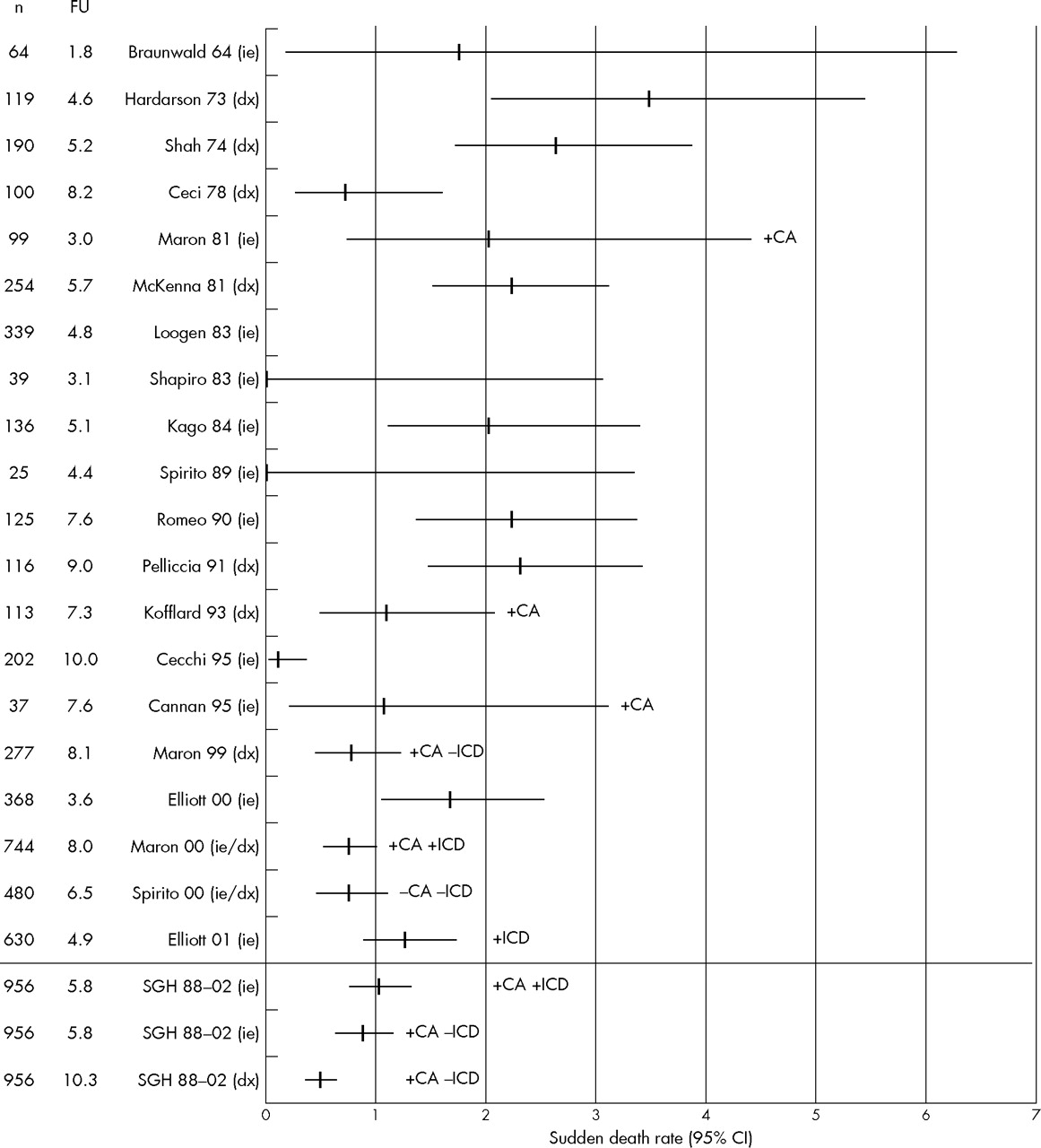
Sudden death rates in published natural history studies of hypertrophic cardiomyopathy. −CA, excluded resuscitated cardiac arrest; +CA, included resuscitated cardiac arrest; CI, confidence interval; dx, follow up started from diagnosis of hypertrophic cardiomyopathy; FU, follow up (years); ie, follow up started from initial evaluation; −ICD, excluded implanted cardioverter-defibrillator (ICD) discharge; +ICD, included ICD discharge; n, number of patients studied; SGH, St George’s Hospital.

{kind=link}
{kind=link}
{kind=link}
All cause death rates in published natural history studies of hypertrophic cardiomyopathy.
All cause mortality over the last 10 years was also lower (median 2.5% (range 0.8–3.6%) v median 2.9% (range 0–6.0%)). For seven studies, it was possible to calculate sudden death rates with and without resuscitated cardiac arrests and appropriate ICD discharges as end points (table 3). For our own institution, this analysis resulted in an annual sudden death rate that ranged from 0.4% to 1.0%.
Annual sudden cardiac death rates together with their 95% confidence intervals (CIs) from seven selected natural history studies in which calculation of different rates was possible depending on inclusion or exclusion of resuscitated cardiac arrest (CA) and appropriate ICD discharges as end points
DISCUSSION
This study shows that published survival rates of patients with HCM have improved from the 1960s onwards, with a reduction in all cause annual mortality from 5% or more to less than 3%, and a fall in annual sudden death mortality from 3% to 1%.
The narrow range of death rates in reports published over the past 10 years suggests that referral bias is less important in the modern era, indicating that common management protocols such as the joint American College of Cardiology/European Society of Cardiology guidelines are applicable to all patients with HCM, irrespective of the setting in which care is delivered.10
Causes of death in HCM
In our cohort, sudden cardiac death was the most common mode of death with an annual incidence of less than 1%. Patients who did not suffer sudden cardiac death had a generally benign clinical course, the major adverse outcomes being death from heart failure, cardiac transplantation, and stroke. Fatal cerebrovascular accidents were uncommon in this series but were associated with atrial fibrillation, emphasising the importance of prophylactic anticoagulation and the management of atrial arrhythmia.
Some patients with HCM are at risk of infective endocarditis and it was the practice throughout this study to recommend prophylactic antibiotics to patients with left ventricular outflow tract obstruction or a murmur. The annual rate of infective endocarditis was comparable with that reported recently.33
Impact of referral bias
Given the retrospective nature of this analysis, it is impossible to provide definitive reasons for the change in survival rates seen over the past 40 years. Changing practice in many countries has meant that increasing numbers of mildly affected patients are identified through family screening (25% in this cohort). With few exceptions, however, the proportion of patients with HCM diagnosed in this way is not reported in published survival studies and the effect of this lack on published survival rates could not be assessed. Some of the patients in our own cohort did not fulfil conventional diagnostic criteria for probands in that they had a maximum left ventricular wall thickness less than 15 mm; their inclusion is this study reflects current practice of using published diagnostic criteria for relatives of patients with an unequivocal diagnosis.12 These patients might have contributed to the low mortality in our series, but as we and others have shown, many relatives with lesser degrees of hypertrophy can die suddenly or have severe restrictive physiology.34,35 Lastly, advances in disease management such as ICDs and the prevention of thromboembolism in patients with atrial fibrillation are likely contributors to the lower event rates.
When we examined data from the past decade, we found some discernable differences between the patients followed up at our unit and those seen at some other centres. In particular, patients tended to be younger and had a higher incidence of familial HCM and family history of sudden cardiac death, a reflection, perhaps, of the systematic approach to the evaluation of families at our institution. Despite these differences, however, the survival of patients in our institution was very similar to that reported from other centres. Moreover, the small differences that did exist between our own cohort and other recent studies can be accounted for by variation in statistical methods. In particular, sudden death rates were higher in studies that included ICD discharges and resuscitated cardiac arrests as sudden death equivalents.
Clinical implications
Given the relative rarity of HCM, only research groups with a particular interest in the diagnosis and management of the disease are able to recruit a sufficient number of patients to conduct survival studies. This makes a degree of selection bias inevitable in anything other than a large scale community based epidemiological survey conducted over several decades. Nevertheless, this study suggests that, irrespective of the referral source, the natural history of most patients with HCM is relatively benign if those at risk of sudden cardiac death and other complications are identified and treated appropriately.
REFERENCES
Supplementary materials
The table is available as a downloadable PDF (printer friendly file).
If you do not have Adobe Reader installed on your computer,
you can download this free-of-charge, please Click hereFiles in this Data Supplement:
- [view PDF] - Table 2. Comparisons of clinical features and outcomes in selected natural history studies.
Footnotes
-
Published Online First 10 October 2005